QbD理念的誕生
QbD,即品質源于設計(Quality by Design),起源于20世紀70年代豐田公司為提高汽車品質而提出的創造性概念,通過在通信、航空等領域的發展逐漸形成。對于制藥業來說,這一概念最早出現在ICH Q8中:“一種系統的開發方法,始于預先確定的目標、產品和過程的理解和進行。”Ess控制基于完善的科學和質量風險管理,是基于充分的科學知識和質量風險管理的系統化研發方法。從預定的目標出發,強調對產品和過程的理解和過程控制”。2005年,FDA開始積極倡導和推廣FDA。
QbT理念與QbD理念
QbT(質量源于檢測)
QB代表“通過測試獲得質量”,即質量來自于測試。認為質量是通過測試注入到產品中的,產品的質量取決于最終的測試。其基本控制框架見下圖1。
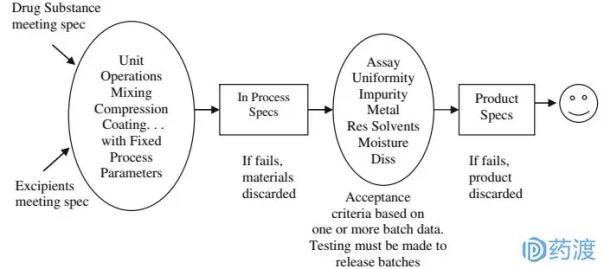
圖1:1:QbT系統控制質量框架圖
在該系統中,產品質量保證主要通過以下步驟完成:
原材料檢測;
活性成分的生產;
固定制劑生產技術;
中間體(過程中的物質)的檢測;
最終產品檢驗。
其中,起始原料的檢測主要包括原料(有效成分)和輔料的檢測。一般來說,這些原料只有滿足生產商、FDA或其他標準(如USP)的要求才能被采用,但一般不會被進一步認可。因此,廠商為了降低風險,一般會制定更嚴格的內控標準,但嚴格標準的具體來源自己可能并不清楚。所以如果原料稍微晚一點變化,就可能導致制劑廠家。
成品的質量保證也是通過檢測和評估來確定的,看檢測結果是否符合廠家和FDA的標準要求。如果沒有,藥品就報廢了;而失敗的根本原因也一直沒有完全搞清楚。出現一批故障后,廠家在不了解故障原因的情況下,仍然冒險繼續生產,直到找到故障根源并解決;或者FDA批準修訂相關可接受標準的補充申請(如放寬部分標準限值),同時對之前“不合格”的批次進行重新合格。
同時,為了保證制劑中間體符合FDA批準的相關標準,生產企業要對大量中間體進行檢測(如混合均勻度BU、片劑硬度)。制造商不得隨意更改批記錄中的生產參數,也不得更改任何生產工藝,除非已向FDA發起變更補充申請并獲得FDA批準;由于這個原因,FDA面臨著大量的CMC補充申請變更。例如,從2005年到2006年,FDA仿制藥辦公室收到了3000多份CMC補充申請。
在QbT概念下,許多參數是固定的。固定的(或不靈活的)生產工藝和大量的測試樣品是保證產品質量的關鍵手段,同時也要求生產工藝的一致性。總之,由于整個R&D方案和生產方案的認知度不高,出現了一套硬性的、不靈活的可接受標準,很多產品本身符合臨床要求,卻因為這個硬性的質量標準而被禁止放行。或者廠家選擇向FDA發起補充申請,修改相關標準。另一方面,生產廠家對其產品所用的原輔材料了解不多,對生產工藝也不太了解,無法識別哪些性能或參數或步驟是影響產品質量的關鍵因素。但是這些關鍵信息無法正確傳遞給FDA的審評人員,所以FDA的審評人員只能采取保守的方式。
鑒于這種監管模式,所有的產品質量保證似乎都只與檢測標準有關,而沒有考慮到消費者的風險;這種監管模式導致生產廠家和審查人員在低風險產品上花費了過多的時間和資源,而對于風險相對較高的產品卻沒有足夠的時間和資源。可以說,這種監管模式的結果是所有產品的資源平均分配,而實際上有些產品可能不需要那么多資源,而有些產品則需要更多資源。比如,對于審評者來說,他們認為對于劑型相對復雜的產品(如控釋制劑、透皮制劑)或治療窗較窄的產品,與其他劑型(如口服固體速釋制劑)沒有明顯區別。事實上,劑型復雜的產品或者治療窗窄的產品,在某些方面可能需要更深入的研究,而普通產品可能不需要花太多時間去研究影響不大的地方。
綜上所述,傳統的產品質量保證模式歸因于生產過程的不靈活和對產品檢測和控制的依賴;但是,在保證產品質量的前提下,如何提高產品的有效性和產品生產的效率,卻很少或根本沒有得到重視。因此,對于工藝或劑型復雜的產品,無法很好地識別放大過程中的關鍵點。產品標準也是通過一批或多批數據檢測建立的。
QbD(質量源于設計)
ICH Q8指出,產品的質量不能由檢驗給出,而是由設計給出。了解這一點非常重要。ICH Q6A強調質量標準“規范”的作用:質量標準是一項重要的質量指示,由制造商提出并論證,經管理機構批準,作為批準產品的依據。所以,起決定作用的是生產廠家,而不是FDA批準。
QbD是一種基于風險的系統、科學、全面、主動的藥物開發方法。從產品概念到產業化都是精心設計的,是對產品屬性、生產工藝、產品性能之間關系的透徹理解。這意味著從藥物研發的開始階段就要考慮最終產品的質量,包括處方設計和開發、生產工藝路線的選擇和確定等。QbD是從患者的角度識別產品的關鍵質量屬性,并將其轉化為藥物研發和生產的控制因素,主要包括以下步驟:
確定目標藥物質量概況的QTPP
設計開發處方和生產工藝,并系統評估、了解和改進。
識別關鍵質量屬性CQAs、過程參數和變量來源。
結合風險管理制定適當的控制策略。
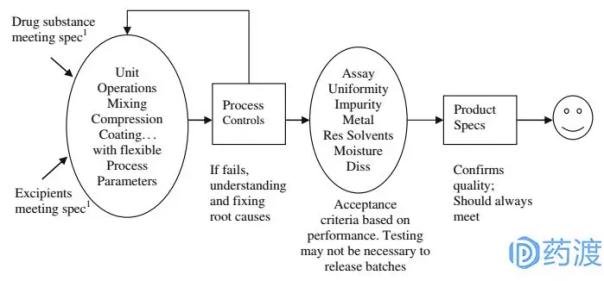
圖2: QBD框架圖
圖2是QbD的框架圖。與QbR不同,QbD認為產品質量標準的建立應結合臨床相關性和產品性能,對生產工藝和產品質量標準進行提前設計和控制。藥品質量的保證是一個系統的研究方法,依賴于對處方工藝的理解和控制,而最終產品檢驗只是保證產品質量的一致性,而不是生產工藝或過程控制的一致性。因此,基于QbD的標準總體上是合理的、嚴格可控的。
目前我們可以接受的內容設置標準和QbD的思路很像。一般我們通常把產品含量的可接受標準定在90%-110%,個別產品因為臨床原因定的標準是95%-105%。這個標準不會因為實際測試數據而改變:即使大部分測試數據都集中在98%-100%,我們還是會把含量的可接受標準定在98%。然而,另一方面,我們似乎根據QbT概念設定了可接受的解散標準。在溶出度標準的制定過程中,我們主要基于實驗室批次數據或小批量數據,試圖找到具有分辨力的溶出曲線,而忽略(或不太關注)溶出曲線與藥物體內行為的相關性。根據QbD的概念,溶出曲線的建立應盡可能反映藥物在體內的行為。對于BCS和口服固體速釋制劑,體內吸收限制步驟取決于藥物的吸收過程,而具有快速溶出特性的藥物不是體內吸收限制步驟。因此,他們的驗收標準可以比實際測試數據更寬。同樣,對于BCS II和IV藥物,需要仔細選擇盡可能反映內部環境的溶出條件和可接受的標準。
雜質(或降解產物)的可接受標準是藥物的另一個CQAs,安全性是其標準建立的底線。根據QbD概念,雜質的可接受標準應該是基于其定量極限或生物安全級別(四個級別,其中I級危害最小),而不是基于實際檢測得到的實驗記錄或批次記錄。一些雜質的可接受標準也可以通過參考RLD制劑來建立(參考文章ANDA:根據FDA考慮雜質的要求)。
據悉,為了申報海外注冊,上海醫藥集團新一醫藥公司已經在兩個項目中使用了QbD。資料顯示,傳統方法無法穩定控制項目質量,一次通過率低。采用QbD后,50批次產品一次合格率100%。因此,從長遠來看,實施QbD可以有效提高藥品質量的合格率和穩定性,從而降低生產成本。所以,QbD的理想狀態是一個雙贏的結果:對于生產者來說,可以減少監管壓力,降低生產成本;對于監管者來說,可以在不犧牲質量的情況下減輕監管壓力;對于患者來說,可以獲得有效的藥物,更好的保證產品質量。
小結
在藥物研發和建立質量標準的過程中,QbT和QbD遵循完全不同的理念:QbT理念下的產品質量標準是基于一系列小批量數據。如果這些數據是可接受的,則應為其制定相應的可接受標準,后續每個生產批次的檢測結果都應符合這些標準,從而通過合格的檢測結果來保證最終產品質量與生產工藝的一致性;在QB理念下建立的質量標準并不要求每一批都要檢測,因為它的標準是建立在對處方工藝的理解或工藝控制的充分證明的基礎上,更加靈活。
QbD的實施需要企業對生產流程進行深入的研究,同時也需要在硬件上進行一定的投入。但目前國內的企業都在“趕審批”,沒有時間、精力和資金進行如此細致深入的研究。看起來他們沒有QbD也能活,但從長遠和發展的角度來看,使用QbD的企業可以走得更遠。
參考資料
1.ICH Q6A的中英文版本
2.ICH Q8的中英文版本
3.制藥質量的設計:產品和工藝開發,理解和控制
4.QbD被認為是一種國際商業機會渠道,受到FDA的積極推動。